【P#14】医薬品の開発(2)〜TLOと事前相談費
今回は、開発の流れの中で、候補の化合物の選定(研究)と実生活に役立つようにデータを集める、という2つのポイントからより医薬品の開発の詳細に迫っていきたいと思う。

一つ目は、研究について。
米国では、1980年代に入り、政府の補助金によって行われた基礎研究を製品に転換、技術移転をスピードアップする法律が次から次へと成立させていった。その中でもっとも重要なものは、バーチ・バイ(Birch Bayh)上院議員とロバート・ドール(Robert Dole)上院議員によって起案されたバイ・ドール法(Bayh-Dole act:Patent and trade mark act amendments of 1980:アメリカ合衆国商標法修正事項)だ。

かつて、米国国立衛生研究所(NIH(National Institute of Health))から得た助成金で行った研究成果の特許はすべて政府に帰属していたが、この法律によって大学やベンチャーなどが特許をとることが出来るようになった。今まで、税金で行われた研究の成果は公共(=国)のものと規定されていたのが、大学が助成金で行われた研究による成果に対して自ら特許をとり、ライセンスを供与し、特許使用料を取れるように変わったと言っていい。
製薬会社が国の税金によって行われた研究特許を商売道具として使っていいのか?という議論もあるが、イノベーションを生み出す一つの大きなきっかけになったのは確かだ。ちなみに日本版バイ・ドール法は、産学活力再生特別措置法(1999年施行・2003年改正)という形で成立している。

この法律のため、当時台頭していたバイオテクノロジーのベンチャー企業の急増や製薬企業の巨大化が促進される。現在、大学研究者が自らの研究成果を実用化するために設立した会社が新薬の開発の初期段階を担っている。そして、成果をあげると、製薬会社との巨額な取引を行うことになる。大学側でも、技術移転事務局(TLO(Technology Licensing Organization))というインフラが整備された。ということは、製薬企業はリスクの高い新薬開発のために自ら研究を行う必要がなくなり、こうしたベンチャー企業やNIHなどに依存するようになってきている。
私が世界一周前にいたBiogen Idecは、MITやハーバード大学が集中するマサチューセッツにあり、ベンチャーや大学から上がってくる成果を買い取り、それを開発していくという目利きに長けていた。
二つ目は、開発と当局について。
臨床試験は3段階に分かれており、健常人で安全性を調査するPhase 1、実際の患者に投与して投与量をきめるPhase 2、実際に既存の治療法を比較して薬の効果と安全性を比較するPhase 3について触れた(【製薬コラムVol.3】参照)。
その際に、治験を行う際にはPMDAへの事前相談、治験届の提出、治験実施計画書(Protocol)等の書類の提出が必要だが、PMDAへの事前相談ややり取りは年々スピードが上がっている(平成19年度が通常品目の平均審査日数が20.7ヶ月から平成23年度が同日数11.5ヶ月に飛躍的に改善(PMDA資料)
実は、米国審査当局の米国食品医薬局(FDA、Food and Drug Administration)は、製薬業界からの圧力によって、製薬企業からお金を受け取っていてよいとする法が成立。1992年に、処方薬審査料法(Prescription Drug User Fee Amendment)が成立した。実は、FDAの医薬品評価研究センター(CDER(Center for Drug Evaluation and Research)の約半分を審査料は占めるという。

この法律は5年ごとに改定され、2008年に調べた際に、医薬品承認審査料は1品目につき57万6千ドル(約7,000万円)だった。 この審査料のわずかな額が安全モニタリングに使われているに過ぎず、大部分は医薬品承認プロセスのスピードアップに使われる。この法律が決まってから、FDAは新薬承認申請の審査のためにFDAは1000人がスピードアップのために増員された。FDAは他に重要性のある医薬品製造基準の遵守状況の監視、医薬品市場の規制といった役割を果たすことや、製薬企業からの影響を排除できないため、当局としての中立性が損なわれるといった懸念が出ている。
現に、審査が加速することによって弊害もでている。例えば、発売後に医薬品の添付文書に警告を追記したり、実際に医薬品が市場から撤退するというケースが出てくるようになったことが報告されている(例えば、「Faster Drug Approvals Linked to Safety Issues」参照)。
参考に、FDAの審査の人数は約5,400人。国際比較のため欧州医薬品庁(EMA)とPMDAとの比較も下記に記す。
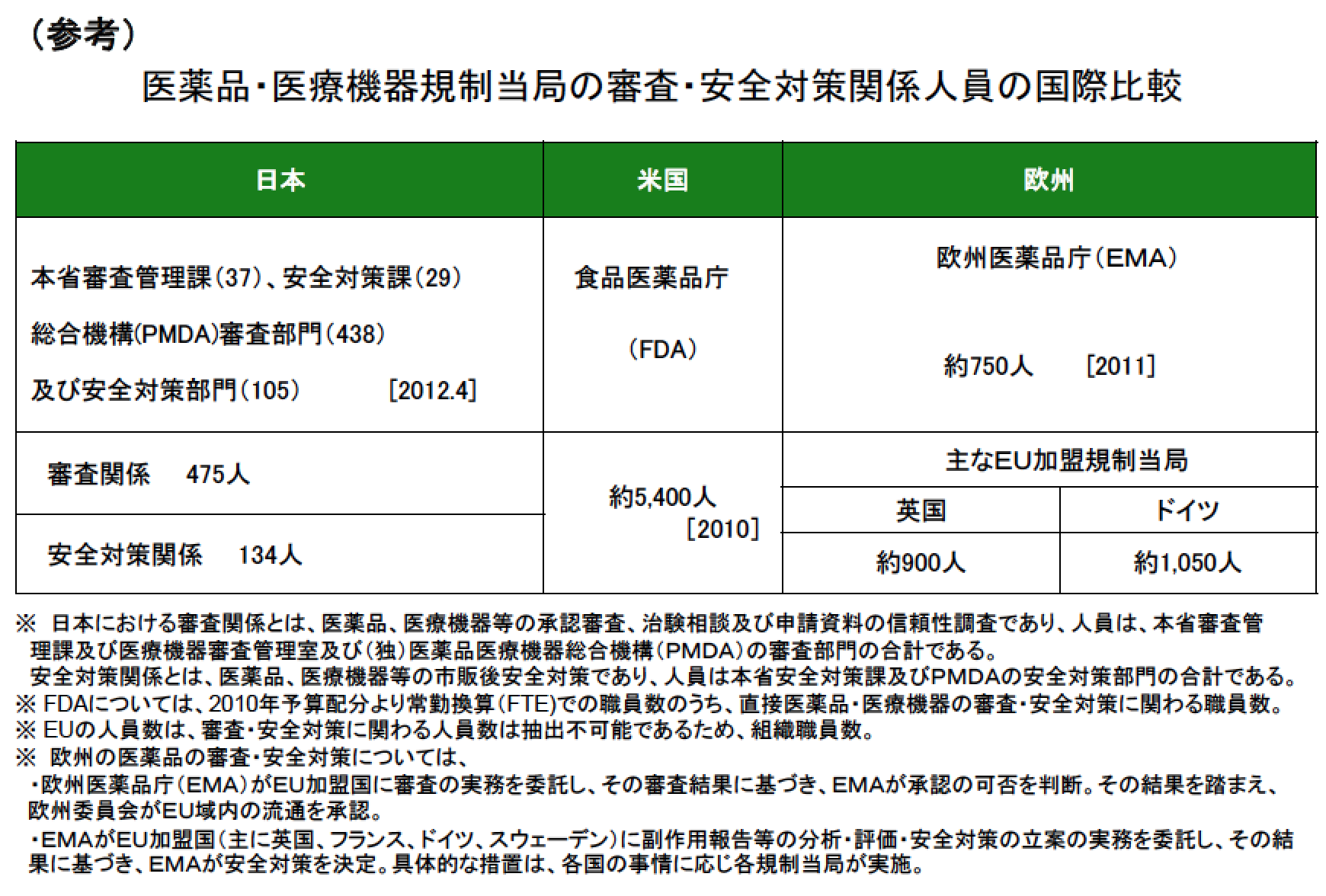
日本も米国の仕組みに従っており、ドラッグ・ラグ(医薬品の開発が諸外国に比べて遅れているという問題のこと)から、製薬企業からお金を取ることで職員を増やしている。
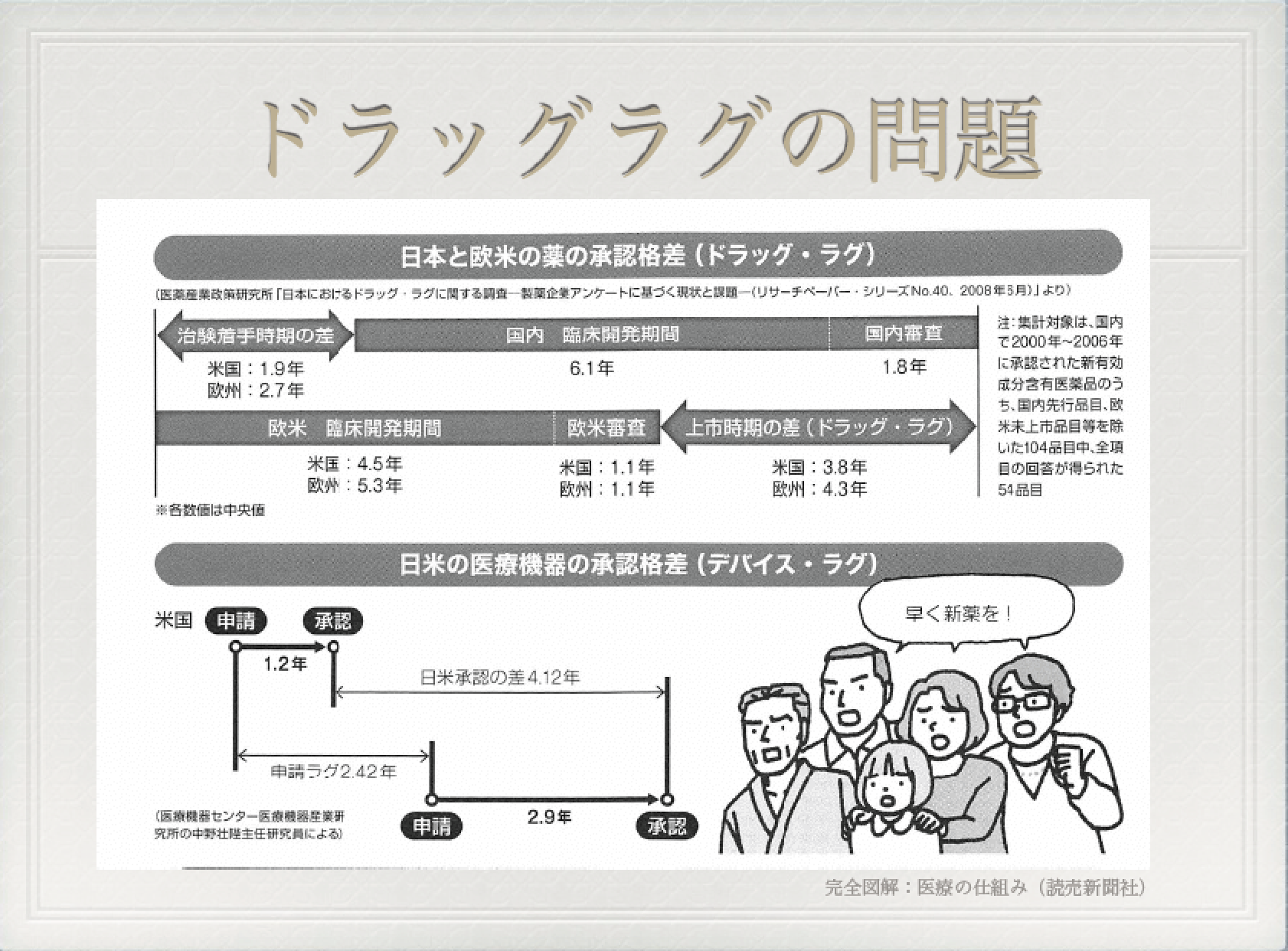
それを財源に、平成30年には1,000人体制を目指すとのこと(平成26年3月31日、PMDAのプレスリリースより)

参考に、製薬会社がPMDAに対し通常の医薬品として支払う相談費用として
医薬品第I相(Phase 1)試験開始前相談は4,360,500円、前期第II相(Phase 2)試験開始前相談は1,669,400円、後期第II相試験開始前相談は3,114,900円、医薬品第II相試験終了後相談は6,183,300円、医薬品申請前相談は6,183,200円となっている(PMDA、手数料の区分参照)。
合算すると、Phase 1〜3と申請前の相談で約2億1500万円かかる計算になる。ちなみに、FDAは、臨床試験の報告書と生データを使って、審査員が独自に解析を進めるのに対して、日欧は、企業に解析・考察を求めそれを審査する。それが結果的に米国は日欧よりも審査の人数が多くなっているのだ。
日欧米の開発、特に製薬会社のお金が審査する際に、入っていることについて簡単にまとめた。
このシリーズをもっと深く読むために
製薬開発の仕組みを知ることは、「薬・エビデンス・健康情報を正しく読む」という大きなテーマの一部です。エビデンスの限界・老化研究の最前線まで含めた全体地図は → 製薬開発・エビデンス・健康長寿の地図
著者:大塚英文(Ph.D.)|渋谷を拠点に、ロルフィング・コーチング・脳活講座を提供。神経科学・哲学・身体知の交差点から、個人と組織の「認識の変容」を扱っている。