【P#24】医薬品の開発(12)〜審査
医薬品として製造し、販売するためにはその製品の「品質(quality)」、「効能・効果(efficacy)」および「安全性(safety)」についてのデータを集め、「製造販売承認」を受けるための審査を受けなければならない。基本的に日本の医薬品の審査・承認は厚生労働省が行うが、米国はFDA(Food and Drug Administration、米国医薬品局)、ヨーロッパはEMEA(European Agency for the Evaluation of Medicinal Products)が審査・承認を行うことになっている。またこれらを含めた医療規制(Regulation)を行う政府機関のことを医療当局(Health authorities:当局ともいう)と呼ぶ。
製薬会社から審査料をとって審査が行わなわれていること(【製薬コラムVol.4】参照)や製薬会社がどのようなチームを組んで申請資料を用意するか(【製薬コラムVol.3】参照)、以前コラムで触れた。
医薬品の品目ごとにその製造販売について厚生労働大臣に承認を受けなければならないが、承認不要のものもある。
例えば、
1)厚生労働大臣が基準に定めている指定する医薬品
調剤用医薬品:病院や町の薬局で調剤をするときに使用するワセリンやパラフィンなど
2)厚生労働大臣が指定する体外診断用医薬品
本来原則的に厚生労働大臣が承認を行うことになっているが、以下の医薬品関して都道府県知事にその権限が委譲されている。
風邪薬、解熱鎮痛薬、鎮咳去痰薬、胃腸薬、瀉下薬、鎮暈薬、眼科用薬、ビタミン主薬製剤、浣腸薬、駆虫薬、鼻炎用点鼻薬、鼻炎用内服薬、外用痔疾薬、みずむし・たむし薬
薬事法では、承認は厚生労働大臣が行うとされているが、その審査については「独立行政法人医薬品医療機器総合機構(Pharmaceuticals and Medical Devices Agency: PMDA)」に委任されている。
今まで、外国で開発された医薬品を日本でも販売できるようになるためには、日本の製薬企業が日本での販売権を購入、承認を取り、国内で製造する場合が多かった。しかし、その後には外資系自身が国内に工場を建て、自ら承認、販売するケースが増えてきた。薬事法の改正により、外資系は外国にいたまま自分で承認を取り、日本の製薬企業などに製造販売させるケースについて、「外国製造医薬品の承認制度」を設けることになった。
医薬品の申請は薬事本部を中心にCTD(Common Technical Document)を準備する。CTDの構成は以下のようになっている。
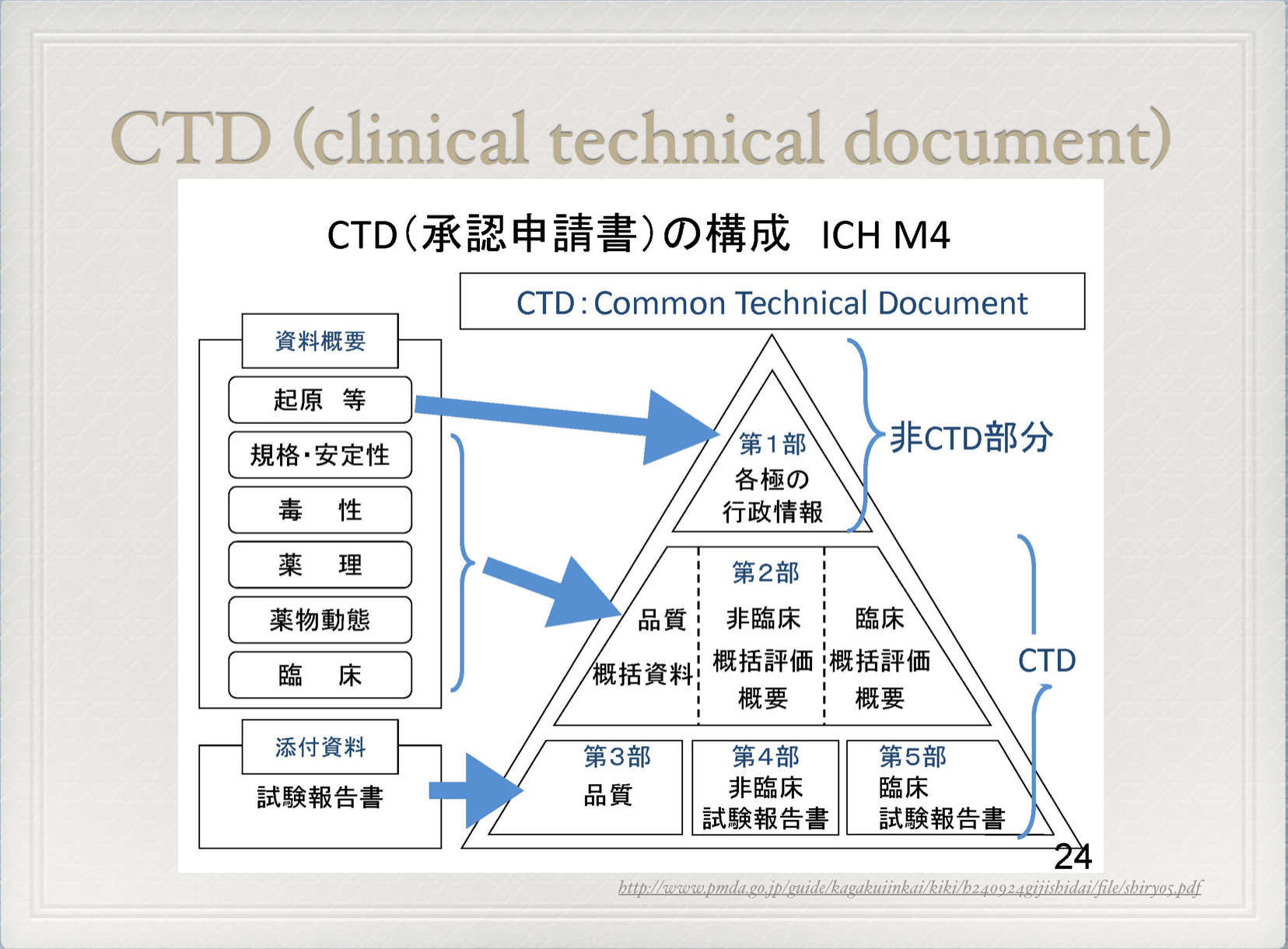
CTDの量はトラック1台分とも言われ、下記の写真を見ていただくとわかるが情報量は膨大だ。

審査の手順については、薬事本部が窓口となりPMDAとのやりとりを行う。

申請資料を提出後、PMDAは照会事項(CTDに関する質問)を用意する。ちなみに、私の携わった医薬品の照会事項は合計で12回。最初200問(副次質問を入れて350問)、その後400問が一斉に送られるという状況もあった。外資系医薬品企業の場合には、照会事項をそのまま翻訳して、外国本社が対応。それを日本語に翻訳するという二重の手間がかかる。 そうは言うものの、照会事項を解凍できるスピードが一つの承認までの律速となるので、製薬会社も必死。ここではスピードが求められる。
医薬品及び医薬部外品の製造管理及び品質管理規則に従っているかどうかをみるために、GMP(Good Manufacturing Practice)の監査が入った後に、医薬品として承認される。
PMDA側は議事録として審査報告書を用意する。この報告書はPMDAのホームページで公開されているが、特許に関わる部分は黒で塗りつぶされている。審査報告書は審査が終わり医薬品の承認されるまえに、PDFフォーマットでPMDAから製薬会社に送付される。審査報告書は、製薬会社の営業戦略を立案するマーケティング本部の必読書とも言われ、医薬品の背景、特に添付文書の根拠となったエビデンスを知る上でよくまとまっている。
医薬品が承認されると厚生労働省と薬価の交渉に移る。医療用医薬品は、薬事法に基づいて承認された後、医療保険の適用を受けるためには、薬価基準への収載手続きが必要とされる。薬価基準収載を希望する製薬企業は所定の申請書類に資料を添えて、新医薬品、報告品目、新キット製品、後発医薬品ごとに定められた期間内に厚生労働省医政局経済課(経済課)へ提出する(薬価の仕組みについては【製薬コラムVol.9】、諸外国との違いについては【製薬コラムVol.10】参照)。

経済課で、製薬企業に対するヒアリングを実施後、申請された品目のリストを作成、保険局医療課(医療課)へ情報を伝達する。医療課では、資料に基づき算定原案を作成、中央社会保健医療協議会(中医協)の下部組織の薬価算定組織(医・歯・薬及び経済学の専門家10人によって構成される組織)で協議、委員の多意見を踏まえて新医薬品の価格を設定する。算定案については、不服があれば再度審議されるが、なければ算定案が決定される。中医協で算定案が承認された後、薬価収載(官報告示)される。
医薬品の薬価収載は時期が決まっていて以下のようになっている。
新医薬品 年4回(承認は原則として60日以内、遅くとも90日以内)
キット品目 年2回(6月及び12月を標準)
後発医薬品 年1回(3月15日までに承認されたもの:7月収載を標準)
次に、添付文書を中心に語ってみたい。
****************************
医薬品については、時々書いています。下記をご参照ください。
ジェネリック医薬品と先発医薬品の違いは?何を知っていればいいのか?(「医薬品の開発(14)〜ジェネリック医薬品とは?先発医薬品とどう違うのか?値段は?」「医薬品の開発(15)〜ジェネリック医薬品とは?(2)〜どのように作られているのか?法律や品質の問題点について」参照)
医薬品の情報を見極めるためには「疫学」の知識が必要〜疫学とは何か?について(「医薬品の開発(13)〜『疫学』とは何か?〜医療情報をどのように判断するか?」参照)
審査書類は何を用意したらいいのか?製薬会社に勤めた過去の経験をベースに(「医薬品の開発(12)〜審査」参照)
米国の保険制度の特徴の一つはマネージドケア、その特徴について(「医薬品の開発(11)〜マネージドケア」参照)
医薬品は保険を財源としている〜保険の歴史、出来高払いと診療別包括払いの違いについて(「医薬品の開発(9)〜保険の仕組み(1)」「医薬品の開発(10)〜保険の仕組み(2)」参照)
処方箋医薬品の薬の値段(薬価)を決めるために、欧米ではエビデンスを重視するようになった(「医薬品の開発(8)〜薬価(2)〜仕組みと保険制度」参照)
処方箋医薬品は、国によって値段が決まっている〜どのような仕組みになっているのか(「医薬品の開発(7)〜薬価(1)」参照)
医薬品は特許で守られているが、どのような仕組みがあるのか(「医薬品の開発(6)〜特許と後発医薬品」参照)
国の審査が優先的に進む医薬品もある〜希少疾患、オーファン医薬品(「医薬品の開発(5)〜オーファン医薬品」参照)
製薬会社の審査は誰が行うのか?当局(国)の存在(「医薬品の開発(4)〜FDAと審査」参照)
医薬品の開発で何が重要になるのか?主要評価項目について(「医薬品の開発(3)〜エンドポイント」参照)
医薬品の審査には、製薬企業のお金が入っている現実(「医薬品の開発(2)〜TLOと事前相談費」参照)
医薬品の基本的な仕組み(「医薬品の開発(1)〜仕組み」参照)
*****************************