薬はどう審査され、承認されるのか──治験・審査・薬価の全体像

カテゴリ:エビデンスを読む──科学・医療・製薬の構造【第3回】/ 初出:2023年3月 / 更新:2026年
Table of Contents
エビデンスに基づく医療と叫ばれているが・・・
医療では、事実(エビデンス)に基づく治療(EBM、Evidence Based Medicine)が叫ばれている。
EBMによって、製薬会社と医師との共同作業で治療ガイドライン(一種のアルゴリズム)を決定。
医師によって病気が診断されると、治療ガイドラインに沿って、治療が決まる。

前々回のブログでは「エビデンス」の方に力点を置き、
「どのように西洋では知識が生まれるのか?」
治療となる薬は、製薬会社で製造、販売。
これらは「科学」の成果の上に成り立っている。
1980年以降、国の税金で行った研究を大学と研究者が特許を取得できるようになる。
「科学」がビジネスと密接に結びつくようになり、その点を前回のブログにまとめた。

今回は、製薬会社で「薬」を開発する際に、審査はどのように進んでいくのか?
製薬会社が国(審査当局)に支払う「審査費(相談費用)」を中心に、
「審査のプロセスはどのようになっているのか?」
ご紹介できればと思う。
製薬会社の開発の仕組み〜「医療用医薬品」の審査
「薬」は、
薬局で販売されている「市販薬」(一般用医薬品)
と
処方箋抜きでは、販売できない「処方箋医薬品」(医療用医薬品)
の2つがある(薬機法で区分は決まっている)。
今回は、医師の処方箋が必要な医療用医薬品を中心に取り上げていきたい。
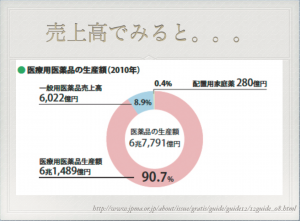
なぜならば、製薬会社から見ると、利益率が高いのは医療用医薬品の方で、市場規模もでかいから。
製薬業界では、製薬の開発本部が医師と共同作業で、薬を
「治験」(治験は、薬機法の用語で、人間で行う臨床試験のことをいう)
と呼ばれる臨床試験を医療機関で行う。
ここで注意なのは、
治験は国が委託した医療機関で行うのではなく、製薬会社が主導して行う
ことだ。
私は、
「治験を製薬会社で行うこと自体、果たして、ニュートラルな結果が得られるのか?」
「医療機関が製薬会社からお金を受け取ることで、臨床試験の結果にバイアスが入る可能性がないのか?」
が気になっているが・・・。
法律(薬機法)で決まっているので、それは突っ込まないことにする。
臨床試験の結果をまとめ、審査当局へ提出する。
(審査当局とは、国の専門部署である「厚生労働省(MHLW)」が審査するが、その業務をPMDA(独立行政法人医薬品医療機器総合機構)に委託する)
何と、治験の結果を含め、薬の承認を得るために、国に提出する書類はトラック1台分にも及ぶのだ。
審査の結果、承認を得て、薬の値段である「薬価」が国によって定められた手順で決まると(つまり薬の値段は国によって決まる)、めでたく発売となる。尚、薬の審査については「審査報告書」という議事録の形でまとまっている(PMDAのホームページで閲覧できる)。

製薬会社で行う試験には、
非臨床(動物)と臨床(人間)=治験
の2つがある。
非臨床で安全性が確認された薬の候補は、
1)健常人で安全性を調査するPhase 1
2)実際の患者に投与して投与量をきめるPhase 2
3)実際に既存の治療法を比較して薬の効果と安全性を比較するPhase 3
の3段階の治験が行われる。
臨床試験の結果を事前に相談する必要性〜「相談費」のからくり
治験を行う際には、さまざまな書類の提出が必要だが、
ポイントとなるのは、
「事前相談」
が必要だということ。
非臨床試験→Phase 1→Phase 2→Phase 3→承認申請
の各段階の前後で、
審査当局と話し合い、議事録に残しておくことで滞りなく、審査が進むように、事前相談をするのだ。
そして
「事前相談」
の各段階で
「相談費用」
を
「製薬会社」から「審査当局」に支払われる。
参考に、
製薬会社がPMDAに対し通常の医薬品として支払う相談費用として
医薬品第I相(Phase 1)試験開始前相談は4,360,500円、
前期第II相(Phase 2)試験開始前相談は1,669,400円、
後期第II相試験開始前相談は3,114,900円、
医薬品第II相試験終了後相談は6,183,300円、
医薬品申請前相談は6,183,200円となっている
(PMDA、手数料の区分参照)。
合算すると、Phase 1〜3と申請前の相談で約2億1500万円かかる計算になる。
ポイントは、製薬会社が、審査当局に相談費を支払うことだ。
「なぜ、このような仕組みになっているのか?」
以下ご紹介させて頂きたい。
審査をスピードアップするプロセスで生まれた「相談費」
国内外では、
「薬の承認が遅い!」
「何とか、最先端の薬を取り入れて欲しい」
といった要望があるのに、
審査が遅いといった課題があった。
これを
「ドラッグ・ラグ」
と呼ぶ。
実は、米国審査当局の米国食品医薬局(FDA、Food and Drug Administration)は、
1992年に製薬企業からお金を受け取っていてよいとする法が成立。
その法律とは、処方薬審査料法(Prescription Drug User Fee Amendment)だ。
何と、FDAの医薬品評価研究センター(CDER(Center for Drug Evaluation and Research)の約半分を審査料は占めるに至っている。

法律は5年ごとに改定され、2008年に調べた際に、医薬品承認審査料は1品目につき57万6千ドル(約7,000万円)だった。
この法律が決まってから、FDAは新薬承認申請の審査のためにFDAは1000人がスピードアップのために増員されたと言われている。
FDAは医薬品製造基準を守っているのかを監視する役割、医薬品市場の規制といった役割を果たすことが重要なのだが、
製薬企業からお金が入ることで「利益相反」の影響や、当局としての中立性が損なわれるといった問題が出る可能性が高い。
例えば、発売後に医薬品の添付文書に警告を追記したり、実際に医薬品が市場から撤退するというケースが出てくるようになったことが報告されている(例えば、「Faster Drug Approvals Linked to Safety Issues」参照)。
参考に、FDAの審査の人数は約5,400人。国際比較のため欧州医薬品庁(EMA)とPMDAとの比較も下記に記す。
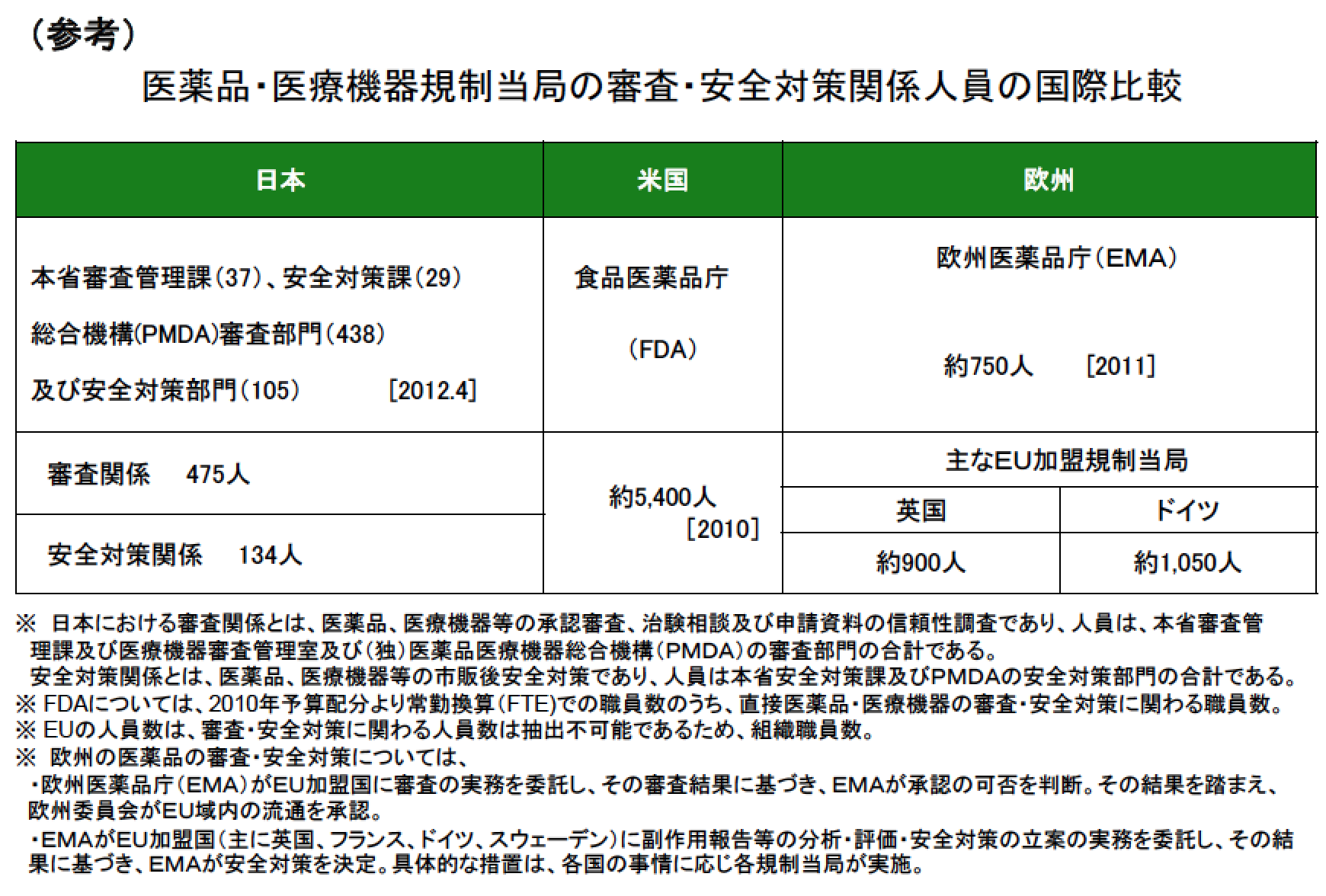
FDAは、臨床試験の報告書と生データを使って、審査員が独自に解析を進める。
対して、日欧は、企業に解析・考察を求めそれを審査する。
それが結果的に米国は日欧よりも審査の人数が多くなっているのだ。
日本も米国の仕組みに従っており、ドラッグ・ラグ(医薬品の開発が諸外国に比べて遅れているという問題のこと)から、
製薬企業から審査費を徴収することで、人件費の当て、審査のスピードアップを図っている。
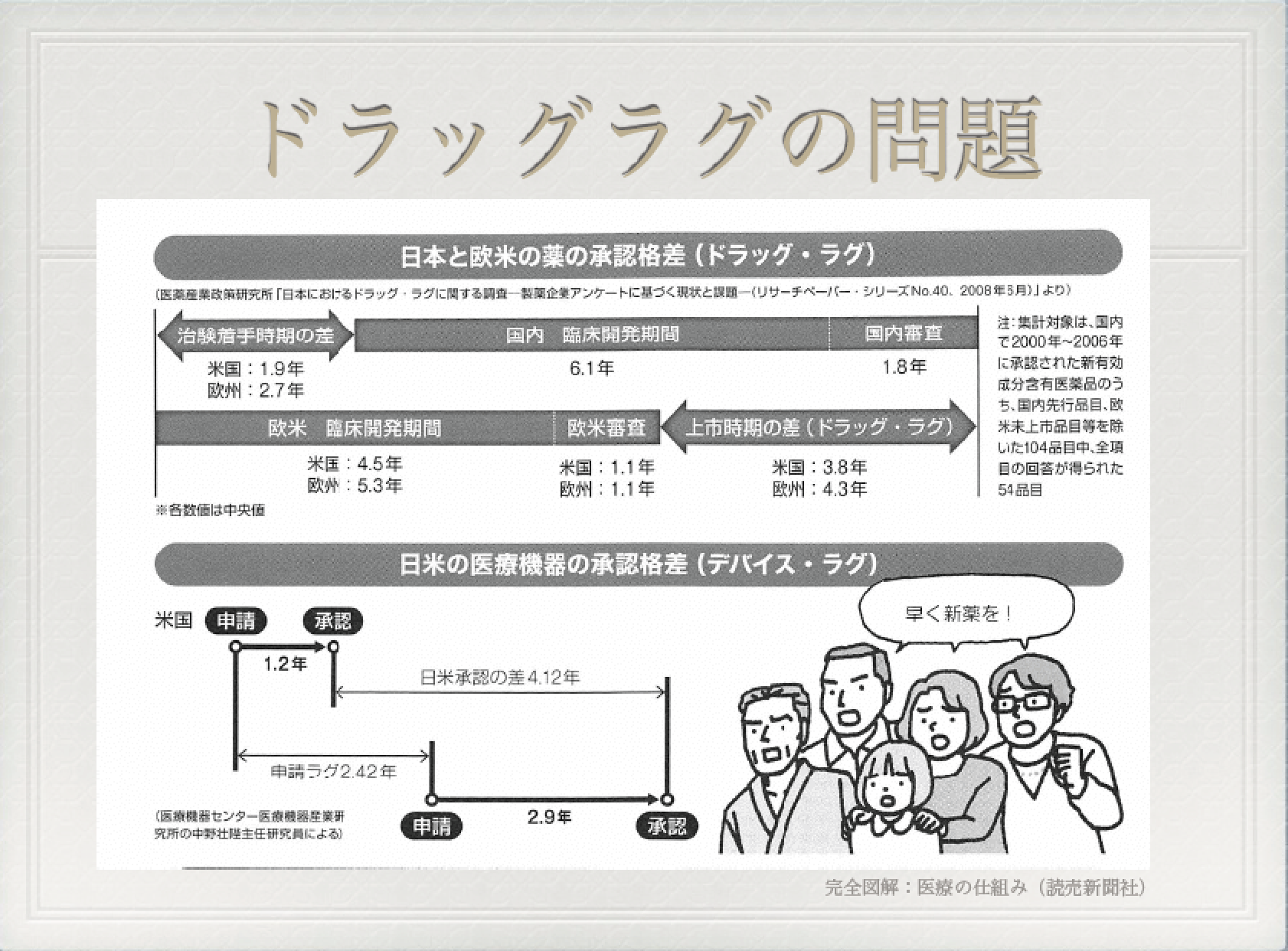
実際、審査費を使って、平成30年には1,000人体制を目指すとのことが審査当局の中で発表されている(平成26年3月31日、PMDAのプレスリリースより)。

このようにして、平成19年度が通常品目の平均審査日数が20.7ヶ月から平成23年度が同日数11.5ヶ月に飛躍的に改善していった(PMDA資料)。
まとめ
今回、製薬会社が主導となって、治験を進めていくプロセスで、審査当局がどう関わっているのか?
を中心にまとめさせていただいた。
西洋医学は、ビジネスと結びつくことで、最新の治療法や薬が普及していったというのは、事実。
が、そのプロセスがどうなっているのか?を知ることがEBMを知る早道だと思っている。
少しでも、この投稿が皆さんにお役に立てれば幸いです。
このシリーズをもっと深く読むために
エビデンスの構造を知ることは、「薬・健康情報を正しく読む」という大きなテーマの一部です。製薬開発の仕組み・老化研究の最前線まで含めた全体地図は → 製薬開発・エビデンス・健康長寿の地図
関連記事
| シリーズ | 記事 | つながり |
|---|---|---|
| 認知バイアス【理論編】 | 第4回:バグより見えにくいノイズ | 査読・臨床判断のノイズ問題と同じ構造 |
| 認知バイアス【理論編】 | 第2回:バグの正体 | 確証バイアスとエビデンスへの過信 |
| 健康・長寿 | ラパマイシン──太平洋の火山が生んだ長寿の鍵 | エビデンスを知った上での老化研究の読み方 |
| 西洋医学 | 製薬開発・エビデンス・健康長寿の地図 | 3層の全体地図 |
著者:大塚英文(Ph.D.)|渋谷を拠点に、ロルフィング・コーチング・脳活講座を提供。神経科学・哲学・身体知の交差点から、個人と組織の「認識の変容」を扱っている。